|
1.巁壔僂儔儞偺擬揱摫棪(MD)
丂妀擱椏儁儗僢僩偲偟偰梡偄傜傟傞巁壔僂儔儞傗MOX偺擬揱摫棪偺應掕偼崲擄偱偁傝丄娤應抣偼悢寘偺僶儔偮偒偑偁傞丅偙偺傛偆側崲擄側忬嫷壓偱暘巕摦椡妛(MD)偺庤朄偵傛傞擬揱摫棪偺寁嶼曽朄偺妋棫偵戝偒側婜懸偑婑偣傜傟傞偺偼昁慠偱偁傞丅偟偐偟側偑傜丄擬揱摫棪摍偺桝憲學悢偺寁嶼偼丄斾擬摍偲摨條宯偺擬暯峵忬懺偺備傜偓偵婎場偟偨暔棟検偱偁傝丄婎杮揑偵偼宯偺旕暯峵忬懺傪庢傝埖偆栤戣偱偁傞偺偱丄偙偲偼娙扨偱側偄丅
丂杮尋媶偱偼MD偺寁嶼忋媅帡揑側旕暯峵忬懺傪幚尰偝偣丄忋婰偺栤戣傪夞旔偟丄岠棪傛偔擬揱摫棪傪寁嶼偡傞偙偲偵惉岟偟偨傕偺偱偁傞丅乮廔椆乯
|
|
|
2.婡擻惈僇儔乕僼傿儖儉偺暘巕愝寁(MC, MD)
丂杮尋媶偼丄壗搙偱傕婰榐偟偨傝徚嫀偟偨傝偡傞偙偲偑壜擻側僇儔乕僼傿儖儉偺暘巕愝寁偺婎杮宍傪扵嵏偡傞偙偲傪栚揑偲偟偨傕偺偱偁傞丅偙偺宯偼斾妑揑寴偄朹忬偺崅暘巕偲愼椏偺暘巕偐傜側傞擇惉暘宯偐傜側傞偑丄愼椏偼敪岝偺儊僇僯僘儉偵偼寚偐偣側偄廳梫側梫慺偱偁傞傕偺偺丄宯慡懱偺峔憿媦傃僟僀僫儈僢僋僗傪寛掕偡傞忋偱偼傓偟傠儅僀僫乕側栶妱傪峴偆偙偲偵夁偓側偄乮偲偄偆壖掕偺尦偱寁嶼傪幚峴乯丅
丂朹忬偺崅暘巕偺摢晹暘偲偦傟埲奜偺晹暘偲偼堎側傞尨巕孮偐傜側傝丄廬偭偰偙傟傜偺憡屳嶌梡偺憡堘偑宯偺拋彉偲柍拋彉峔憿傪巟攝偡傞廳梫側僷儔儊乕僞乕偲側傞偙偲偑暘偐傞丅杮尋媶偱偼偙傟傜偺憡屳嶌梡偺戝彫偵傛傝惗偢傞宯偺拋彉峔憿媦傃偦傟偵帄傞僟僀僫儈僋僗摍偵偮偄偰徻嵶偵峫嶡偟偨偑丄偙傟偼僇儔乕僼傿儖儉奐敪偺堊偺暘巕愝寁偵廳梫側巜恓傪梌偊傞丅乮廔椆乯
|
|

CR(Color Rewritable)僼傿儖儉偺暘巕摦椡妛僔儈儏儗乕僔儑儞 |
3.傾儖僇儕働僀巁墫僈儔僗偺峔憿偲僟僀僫儈僋僗(MD)
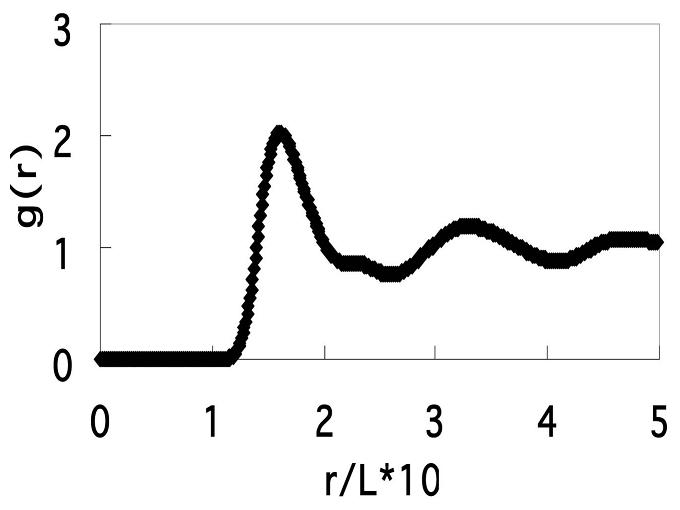
丂Li2SiO3偺僈儔僗忬懺偱偼丄Li僀僆儞偼SiO4偺儐僯僢僩偐傜側傞僱僢僩儚乕僋峔憿偺拞傪偐側傝帺桼偵奼嶶塣摦傪偡傞偙偲偑偱偒傞偑丄偙偺塣摦偺宍懺偼捠忢偺僽儔僂儞塣摦偲偼戝偒偔堎側傞偙偲偑暘偐傞丅枖Li尨巕偺敿暘偺屄悢傪K尨巕偵抲姺偡傞偙偲偵傛傝乮偮傑傝LiKSiO3乯傾儖僇儕僀僆儞偺奼嶶偼戝偒偔掅壓偡傞乮崿崌傾儖僇儕岠壥偲偄偆乯丅杮尋媶偼暘巕摦椡妛偺曽朄偵傛傝偙傟傜偺尨場傪扵傞丅
丂偙偙偵庢傝忋偘偨堎忢奼嶶尰徾偼僈儔僗忬懺偵偍偗傞僀僆儞乮尨巕乯偺奼嶶塣摦偵嫟捠偡傞栤戣偱偁傝丄僈儔僗揮堏壏搙傪寛掕偡傞栤戣偲傕棈傫偱偄偰僈儔僗忬懺堦斒偺廳梫側僥乕儅偱偁傞丅杮尋媶偼宲懕偟偰峴偭偰偄傞丅
|
|
儊僞働僀巁儕僠僂儉僈儔僗偺Li僀僆儞偺2懱暘晍娭悢 |
4.乮揹婥乯儊僢僉偺暘巕僔儈儏儗乕僔儑儞(MC,
MD)
丂揹婥儊僢僉偺暘巕儗儀儖偺峫嶡偼傎偲傫偳夝柧偝傟偰偄側偄丅儊僢僉偼屆偔偐傜偁傞媄弍偱偼偁傞偑丄偙傟枠偺儊僢僉媄弍偼mm,
兪m埲壓偺僒僀僘偱梫媮偝傟傞偙偲偼婬偱偁傝丄傓偟傠壜帇僒僀僘偑昗弨偱偁偭偨偲尵偊傞丅偟偐偟側偑傜嵟嬤偺敿摫懱偺婎斦愝寁偵昁梫偲偝傟傞儊僢僉偺媄弍偱偼乣100nm偁傞偄偼偦傟埲壓偺僒僀僘偑巊梡偝傟傞丅偙偺傛偆側僒僀僘壓偱偺儊僢僉媄弍偼枹偩傎偲傫偳枹抦偺悽奅偱偁傝丄幚梡忋傕戝偒側栤戣揰傪偐偐偊偰偄傞丅偙傟傜偺栤戣傪夝寛偡傞嵟傕桳椡乮桞堦丠乯側曽朄偵暘巕僔儈儏儗乕僔儑儞偑偁傞丅nm僒僀僘偱偼奼嶶曽掱幃偺傛偆側楢懕攠幙拞偺塣摦曽掱幃偼傕偼傗惉傝棫偨側偄偲峫偊傞偲丄儊僢僉昞柺傪峔惉偡傞嬥懏丄嬥懏僀僆儞丄梟攠暘巕丄悈暘巕摍傪偡傋偰棻巕偲偟偰庢傝埖偆夝愅曽朄偑梋媀側偔偝傟傞丅
丂杮尋媶偱偼儊僢僉昞柺偺嬥懏僀僆儞偺壔妛斀墳傪儌儞僥僇儖儘(MC)朄傪梡偄偰妋棪揑偵庢傝埖偆丅偙偺慹帇壔儌僨儖傪梡偄傞偙偲偵傛傝寁嶼帪娫偑戝暆偵抁弅壔偝傟傞丅nm僒僀僘偵偍偄偰偼幚嵺偺儊僢僉偱偼揧壛嵻乮梷惂嵻傗懀恑嵻乯傪梡偄傞偑丄暘巕僔儈儏儗乕僔儑儞偵墬偄偰傕揧壛嵻偵憡摉偡傞梋暘偺暘巕傪憓擖偡傞偙偲偵傛傝幚尡宯偲偺憡帡傪峴偆偙偲偑壜擻偱偁傞
丂MC朄埲奜偵傕暘巕摦椡妛僔儈儏儗乕僔儑儞(MD)朄偵傛傝儊僢僉昞柺忋偺僟僀僫儈僢僋僗傪峫嶡拞偱偁傞丅
|
|
儊僢僉偺暘巕僔儈儏儗乕僔儑儞 |
5. NiAl崌嬥偺儅儖僥儞僒僀僩曄懺(MD)
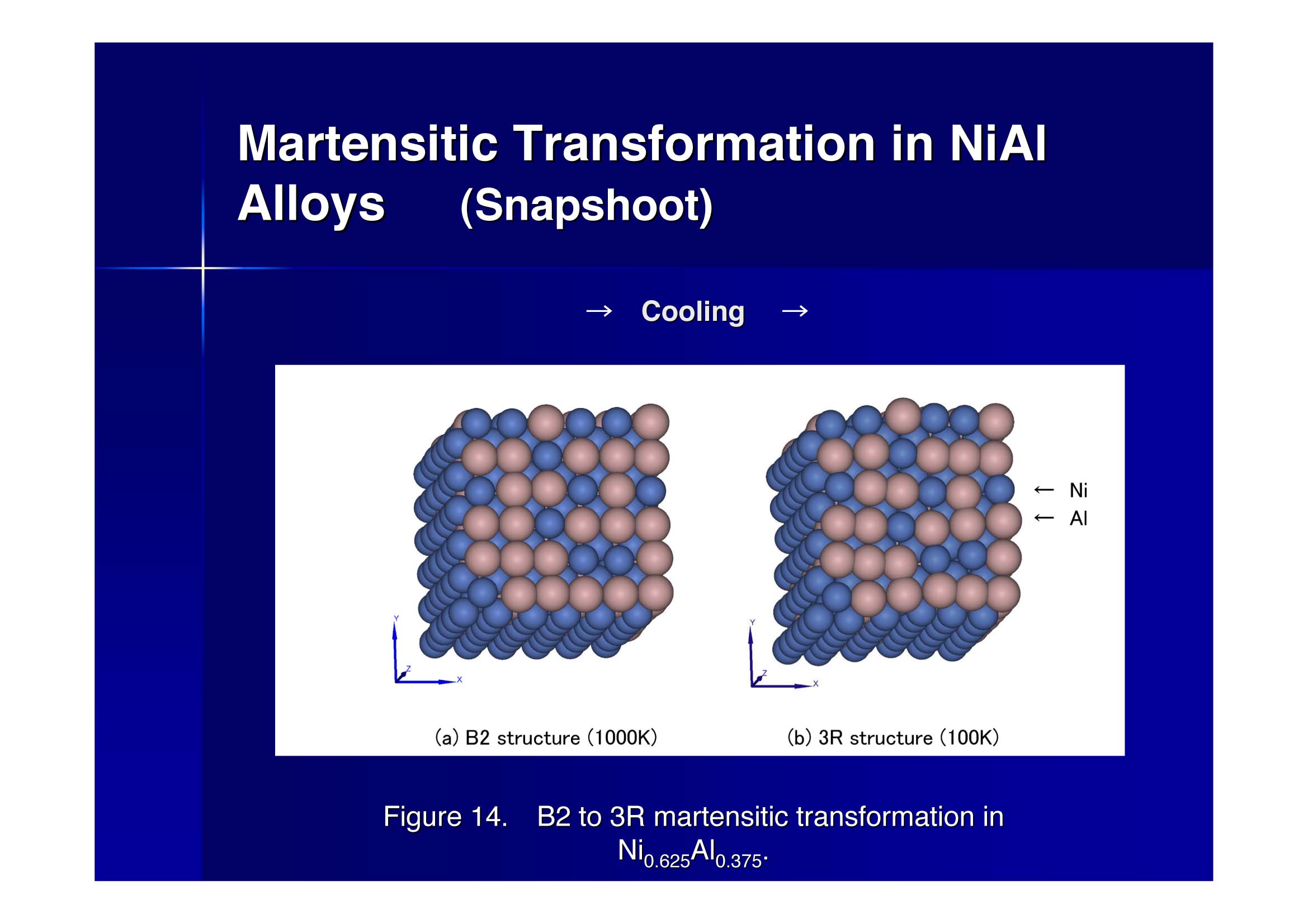
丂崌嬥偺憡曄懺偼嬌傔偰懡條惈傪帵偡丅懡惉暘宯崌嬥偼傕偲傛傝丄擇惉暘宯崌嬥偵偍偄偰傕摨條偱偁傞丅崌嬥宯偺摿挿偼峔惉偡傞嬥懏尨巕偺揹巕揑惈幙傪擛壗偵惓妋偵乮岠棪傛偔乯庢傝擖傟傞偐偵偐偐傢偭偰偄傞丅嵟嬤偺尋媶偱偼僔儏儗乕僨傿儞僈乕曽掱幃傪戞堦尨棟揑偵夝偔庤朄偑偟偽偟偽梡偄傜傟傞偑丄偙偺庤朄偺寚揰偼寁嶼帪娫偑嬌抂偵挿偔側傝丄尷傜傟偨尰幚宯偵偟偐揔梡偱偒側偄偙偲偵偁傞丅
丂杮尋媶偼崌嬥偺儅儖僥儞僒僀僩曄懺偵徟揰傪摉偰偨傕偺偱偁傞偑丄偙偺庬偺栤戣偵戞堦尨棟揑寁嶼庤朄傪揔梡偡傞偺偵偼枹偩帪婜彯憗偱偁傞丅偙傟偵戙偭偰嬥懏丒崌嬥宯偺揹巕忬懺傪岠棪傛偔庢傝擖傟偨曽朄偵EAM偑偁傝丄偙偙偱偼EAM傪奼挘偟偨MEAM傪梡偄偰宍忬婰壇崌嬥偲偟偰傛偔抦傜傟偨NiAl崌嬥偺儅儖僥儞僒僀僩曄懺傪峫嶡偟偨丅僷儔儊乕僞僠儏乕僯儞僌傪峴偆偙偲偐傜丄幚尡偲傎傏堦抳偡傞椙岲側寢壥傪摼偨丅廬棃偺偙偺庬偺寁嶼乮儅儖僥儞僒僀僩曄懺乯偵偼丄帺桼昞柺傪桳偡傞僫僲僒僀僘偺崌嬥傪梡偄偰峴偭偨傕偺偼偁傞偑丄廃婜嫬奅忦審傪梡偄丄僶儖僋壓偱偺幚尡偵懳墳偟偨寁嶼偼弶傔偰偱偁傞丅尰嵼峏偵懠偺崌嬥宯偵傕奼挘偟偰尋媶傪峴偭偰偄傞丅
|
|
NiAl崌嬥偺儅儖僥儞僒僀僩曄懺偺暘巕摦椡妛 |
6.崅暘巕摦揑桿揹摿惈(MD)
丂師悽戙偺捠怣婡婍偺奐敪栚昗偲偟偰悢昐僊僈僿儖僣懷偐傜僥儔僿儖僣懷偺廃攇悢偵巊梡偱偒傞惢昳奐敪偑拲栚偝傟偰偄傞丅偙傟偑壜擻偵側傟偽捠怣検偺旘桇揑側憹戝傪偼偐傞偙偲偑偱偒実懷揹榖側偳偺惈擻偑旘桇揑偵岦忋偡傞丅杮尋媶偱偼偙傟傜偺栚揑偵揔偆崅暘巕桿揹嵽椏偺暘巕僨僓僀儞傪暘巕摦椡妛僔儈儏儗乕僔儑儞偺曽朄偵傛傝峫嶡偟偨傕偺偱偁傞丅暔幙偺桿揹棪偵娭偟偰尵偊偽丄暘巕偺暘嬌乮揹婥憃嬊巕儌乕儊儞僩乯偑尞偲側傞丅崅暘巕嵽椏偺応崌偼暘巕撪偺暘嬌偲暘巕娫暘嬌偲嬫暿偱偒傞偑丄拞偱傕慜幰偺暘晍乮峔憿乯偼変乆偺娭怱偺偁傞廃攇悢懷偺桿揹摿惈偵嵟廳梫偱偁傞丅杮尋媶偱偼偙偺傛偆側暘巕撪暘嬌偺傒傪峫椂偟丄暘巕娫暘嬌偼柍帇偟偰丄揹婥憃嬊巕儌乕儊儞僩偺帺屓憡娭娭悢偺僷儚乕僗儁僋僩儔儉偐傜桿揹摿惈傪媮傔偨丅杮尋媶偼宲懕偟偰峴偭偰偄傞丅
1.乣6.暘巕僔儈儏儗乕僔儑儞偺嶻嬈傊偺墳梡偵偮偄偰乮俈幮偲屄暿偵嶻妛楢実嫟摨尋媶傪幚巤乯
丂丂柤徧丗彠妛婑晅嬥乮俈幮乯
丂丂婜娫丗暯惉俀擭乣
|
|
PPO乮崅暘巕桿揹懱乯偺暘巕僔儈儏儗乕僔儑儞儌僨儖
|
7.崅暘巕丒暘巕廤崌懱偺帺桼僄僱儖僊乕偺寁嶼朄偲挻崅懍愱梡寁嶼僨僶僀僗偺奐敪
丂丂柤徧丗媽捠嶻徣乮NEDO乯
丂丂婜娫丗暯惉俉擭乣暯惉10擭
丂杮尋媶偼丄暯惉俉擭搙乣侾侽擭搙偺俁儢擭偺娫俶俤俢俷採埬宆壽戣尋媶偵嵦戰偝傟幚巤偟偨傕偺偱偁傞丅尋媶偺庡栚昗偼丄暘巕宯偺峔憿揮堏偺尋媶偵偍偄偰幚尡偵捈愙娤應偝傟傞擬椡妛彅検偺拞偱傕帺桼僄僱儖僊乕偼嵟傕廳梫側暔棟検偱偁傞丅偟偐傞偵棟榑偍傛傃寁嶼偱帺桼僄僱儖僊乕傪惓妋偵媮傔傞偙偲偼僄僱儖僊乕側偳偺検偵斾偟偰傕偼側偼偩崲擄側壽戣偺堦偮偱偁傞丅変乆偼杮尋媶偵偍偄偰僋乕儘儞憡屳嶌梡傪娷傓崅暘巕宯偺帺桼僄僱儖僊乕寁嶼傪岠棪偐偮惛搙椙偔寁嶼偡傞寁嶼傾儖僑儕僘儉偍傛傃寁嶼僔僗僥儉乮愱梡暲楍寁嶼僔僗僥儉乯傪奐敪偟偨丅杮寁嶼僔僗僥儉偵傛傝丄埨壙偱偟偐傕悢枩尨巕宯偵偍偄偰僫僲昩掱搙偺婯柾偺暘巕摦椡妛僔儈儏儗乕僔儑儞偑壜擻偱偁傞丅
|
|
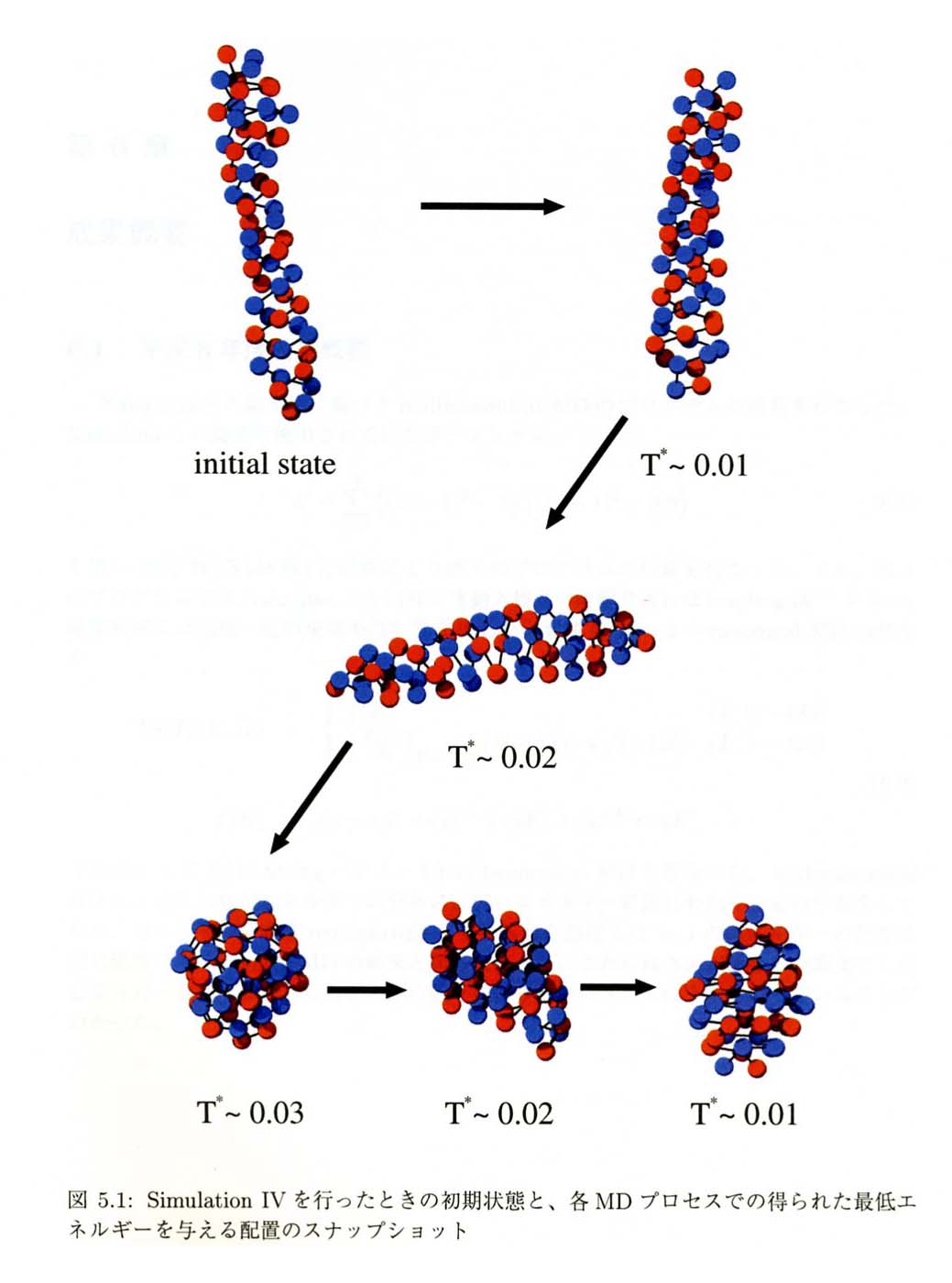
NEDO尋媶奐敪廔椆曬崘彂傛傝揮嵹 |
8. 崅暘巕夛崌乮拋彉乯丒夝棧乮柍拋彉乯偺暘巕榑揑尋媶
丂丂柤徧丗媽壢妛媄弍挕乮JST乯
丂丂婜娫丗暯惉10擭乣暯惉13擭
丂俶俤俢俷巟墖尋媶偱摼傜傟偨惉壥傪敪揥偝偣丄崅暘巕夛崌乮拋彉壔乯丒夝棧乮柍拋彉壔乯偺暘巕儗儀儖偺婡峔夝柧傪栚巜偟偨杮尋媶偼丄俰俽俿乮壢妛媄弍怳嫽帠嬈嵿抍乯寁嶼婡妶梡採埬宆壽戣尋媶偲偟偰嵦戰偝傟丄暯惉侾侽擭侾侽寧乣侾俁擭俋寧枠偺俁儢擭娫尋媶傪峴偭偨傕偺偱偁傞丅杮僾儘僕僃僋僩尋媶偼丄夛崌丒夝棧偺暘巕棟榑揑峫嶡傪巒傔丄暘巕僔儈儏儗乕僔儑儞偵傛傞夛崌柾媅幚尡傪捠偟偰暘巕儗儀儖偺夛崌偵娭偡傞怴偟偄抦尒乮椺偊偽慳悈揑夛崌側偳偺暘巕榑乯傗庬乆偺崅暘巕宯乮抈敀幙側偳乯偺峔憿曄壔乮夛崌丒夝棧乯偺摿幙傪憡屳嶌梡偺宆偍傛傃偦偺戝偒偝偵傛傝夝柧傪帋傒偨傕偺偱偁傞丅偙傟偵傛傝暘巕愝寁傊偺摴昗傪婜懸偡傞傕偺偱偁傞丅
|
|
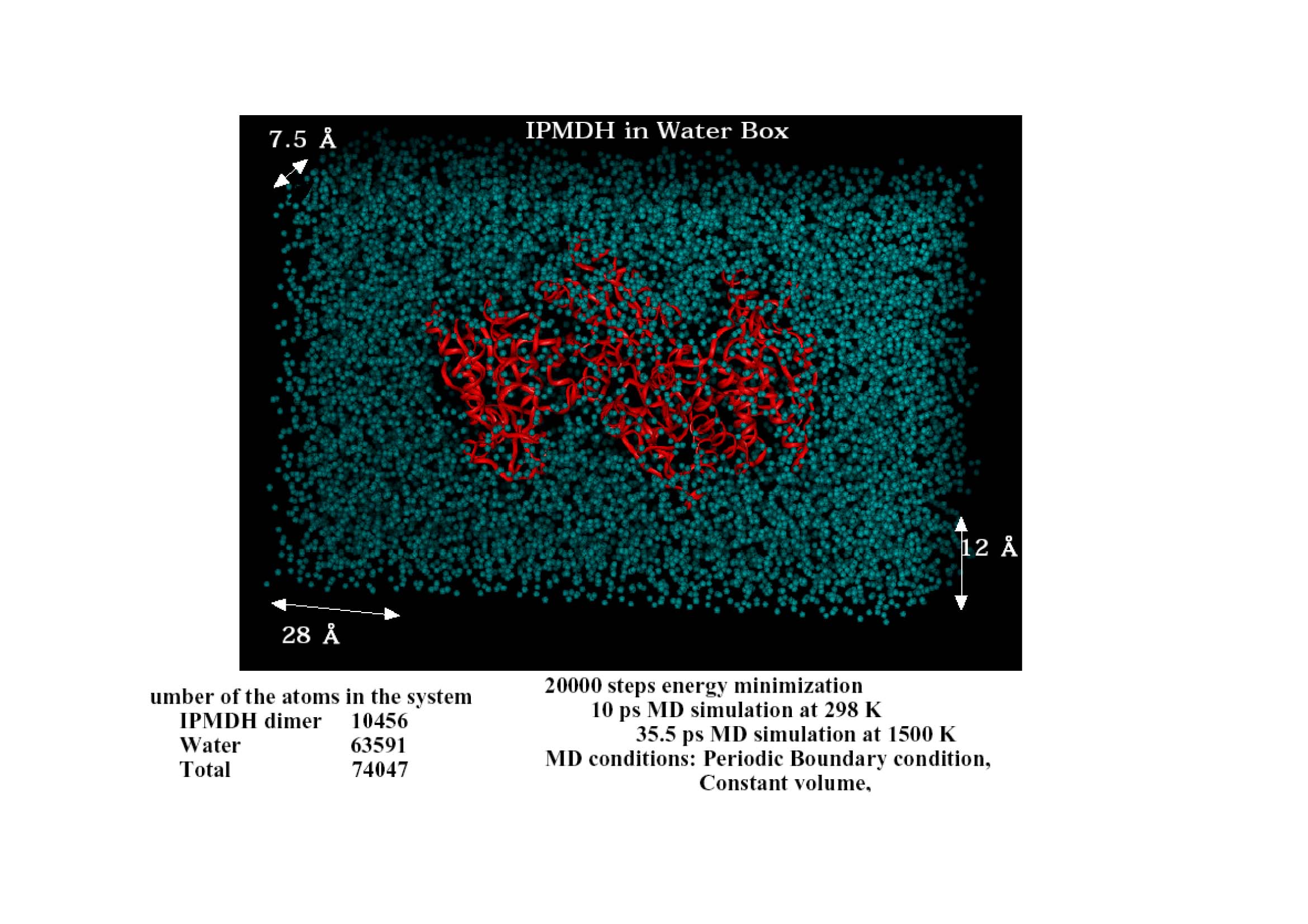
JST尋媶奐敪廔椆敪昞夛乮暯惉14擭3寧乯帒椏傛傝 |
9.暘巕僔儈儏儗乕僔儑儞偵偍偗傞暲楍寁嶼媄朄偺奐敪偲墳梡
丂丂柤徧丗擔杮尨巕椡尋媶強
丂丂婜娫丗暯惉10擭乣暯惉12擭
丂杮尋媶偵傛傝丄屆揟棻巕宯偺暘巕摦椡妛僔儈儏儗乕僔儑儞丒僐乕僪偺暲楍壔偍傛傃揹巕宯僞僀僩僶僀儞僨傿儞僌儌僨儖偺暲楍壔寁嶼僐乕僪偺奐敪尋媶側側偳傪峴側偭偨丅傑偨丄偙傟傜偺暘巕摦椡妛僔儈儏儗乕僔儑儞丒僐乕僪傪嫽枴偁傞暔棟丒壔妛宯偵揔梡傪帋傒偨丅
|
|
|
10.僊僈價僢僩僱僢僩儚乕僋傪棙梡偟偨峀堟壖憐僔儈儏儗乕僔儑儞岺応偺採埬
丂丂柤徧丗捠怣丒曻憲婡峔
丂丂婜娫丗暯惉12擭乣暯惉14擭
丂壖憐僔儈儏儗乕僔儑儞儔儃偺婎斦峔抸偺偨傔僱僢僩儚乕僋愙懕偺婎慴幚尡偍傛傃僸儏乕儅儞僀儞僞乕僼僃僀僗丒棫懱壜帇壔昞尰朄偺婎慴媄弍奐敪傪峴側偭偨丅暘扴幰偲偟偰昁梫側尋媶忣曬廂廤丄僔儈儏儗乕僔儑儞丒僐乕僪偺儌僕儏乕儖壔丄偍傛傃壖憐嬻娫偵偍偗傞棫懱壜帇壔偺奐敪偵嶲夋偟偨丅 |
|
 |
